GIP, also known as gastric inhibitory polypeptide, or glucose-dependent insulinotropic polypeptide, is a 42 amino acid peptide hormone synthesized in and secreted from K cells in the intestinal epithelium. The majority of intestinal K cells are located in the proximal duodenum. GIP secretion is primarily regulated by nutrients, especially fats Intriguingly, bioactive truncated GIP has also been detected in pancreatic a-cells, raising the possibility of an intra-islet a-cell to b-cell connection Glucose-dependent insulinotropic polypeptide (GIP) is expressed in pancreatic islet alpha-cells and promotes insulin secretion Gastroenterology. 2010 May;138(5):1966-75. GIP and its receptor have also been identified in the rodent CNS, including neurons, Schwann cells and oligodendrocytes Glucose-dependent insulinotropic polypeptide (GIP) and its receptor (GIPR): cellular localization, lesion-affected expression, and impaired regenerative axonal growth J Neurosci Res. 2009 Jun;87(8):1858-70.
GIP exhibits potent incretin activity in rodents and human subjects. Unlike GLP-1, which exerts multiple non-incretin activities in the regulation of blood glucose, the primary action of GIP is the stimulation of glucose-dependent insulin secretion. GIP may also play a role in adipocyte biology, however the physiological significance of GIP action in the adipocyte is less well defined. The interaction of GIP with its receptor has been studied at high resolution, demonstrating the importance of hydrophobic interactions for interaction of the GIP alpha helix with the extracellular domain of the GIP receptor as outlined in Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci U S A. 2007 Aug 21; [Epub ahead of print]
To review in more detail the receptor-dependent mechanisms mediating GIP action, see GIP Receptor
The site of GIP expression, the gut endocrine K cell, clearly possesses the molecular machinery required for sensing nutrient intake and secreting GIP following nutrient ingestion. Targeting a modified human insulin transgene to the murine K cell results in appropriate glucose- and meal-related insulin secretion in vivo. See Glucose-dependent insulin release from genetically engineered K cells Science. 2000 Dec 8;290(5498):1959-62. As gut K cells are sparse and difficult to study, there is little known about the molecular biology of the GIP-secreting K cell. Parker and colleagues isolated purified populations of non-immortalized murine K cells from transgenic mice expressing a yellow fluorscent protein under the control of the GIP promoter. Murine K cells were found to express Kir6.2, Sur1, Sglt1, GPR40, GPR119, and GPR120 and GIP secretion was stimulated by glucose, cyclic AMP and linoleic acid as well as tolbutamide but not by modulation of taste receptor activity. GIP+ cells also expressed the proglucagon gene, and at a much higher level, the Pyy gene. See Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia. 2009 Feb;52(2):289-98.
Th importance of the K cell for glucose homeostasis and energy conservation has been studied in mice with targeted transgenic ablation of GIP-producing K cells using a Diphtheria toxin transgene under the control of the rat GIP promoter. Substantial ablation of K cells was achieved and associated with almost complete extinction of GIP expression in the murine small bowel. Unexpectedly, the incretin response was completely lost in these mice, despite normal levels of GLP-1. Moreover, the mice exhibited a partial restoration of the incretin response after high fat feeding yet were resistant to diet-induced obesity, and exhibited a phenotype partially resembling Gipr-/- mice, with increased energy expenditure and enhanced insulin sensitivity Targeted ablation of GIP-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet J Biol Chem. 2008 Apr 17; [Epub ahead of print]
The actions of GIP have been studied by administration of GIP to cells, and infusion in rodents and human subjects. Although classically viewed as an incretin, GIP may also modulate plasma insulin via effects on insulin extraction or clearance in specific experimental conditions. Although GIP infusion in normal human subjects did not affect glucose, insulin, or C-peptide at normoglycemia (5 mM glucose), GIP co-administered with the SU glibenclamide increased plasma insulin but not C-peptide. The mechanisms underlying this intriguing observation remain unclear. See Glucose-dependent insulinotropic hormone potentiates the hypoglycemic effect of glibenclamide in healthy volunteers: evidence for an effect on insulin extraction. J Clin Endocrinol Metab. 2001 May;86(5):2015-9
An important determinant of GIP action is the N-terminal cleavage of the peptide to the inactive GIP (3-42). The enzyme DPP-4, which also cleaves GLP-1 and GLP-2, rapidly inactivates GIP both in vitro and in vivo. See Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. 1993 Jun 15;214(3):829-35 and Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV.Endocrinology. 1995 Aug;136(8):3585-96 and Investigation of glucose-dependent insulinotropic polypeptide-(1-42) and glucagon-like peptide-1-(7-36) degradation in vitro by dipeptidyl peptidase IV using matrix-assisted laser desorption/ionization-time of flight mass spectrometry. A novel kinetic approach J Biol Chem. 1996 Sep 20; 271 (38) :23222-9;
Modification of the GIP N-terminus, such as substitution of D-Ala at position 2, may confer resistance to DPP-4-mediated degradation and result in GIP derivatives with enhanced biological potency. Indeed, [D-Ala2]-GIP1-42 acts a more potent "super agonist" at the GIP receptor in normal rats in vivo when compared to native GIP alone. Furthermore, even greater potency, as assessed by glucose lowering GIP1-42 were not completely correlated with the effects of this molecule on plasma insulin. See Dipeptidyl Peptidase IV-Resistant [D-Ala2]Glucose- Dependent Insulinotropic Polypeptide (GIP) Improves Glucose Tolerance in Normal and Obese Diabetic Rats Diabetes 2002 51: 652-661
A novel degradation-resistant C-terminally truncated GIP analogue, [D-Ala2]-GIP(1-30) was evaluated in a comprehensive series of experiments in rats, mice and cell studies. D-GIP(1-30) robustly improved glucose homeostasis, reduced b-cell apoptosis in islets from VDF rats, preserved b-cell mass in the context of STZ administration, but did not increase body weight in rodents. Intriguingly, D-GIP(1-30) was not as potent as a GIP(1-42) as an inducer of lipoprotein lipase compared to GIP(1-42) in 3T3L1 cells. See A GIP Receptor Agonist Exhibits beta-Cell Anti-Apoptotic Actions in Rat Models of Diabetes Resulting in Improved beta-Cell Function and Glycemic Control PLoS One. 2010 Mar 9;5(3):e9590
Whether enhancement of GIP receptor signaling is a viable strategy for the treatment of Type 2 diabetes remains unclear, as human data demonstrating efficacy of GIP agonists in the setting of Type 2 diabetes is quite limited, as discussed below.
Consistent with the role of DPP-4 in the degradation of GIP, levels of intact GIP (1-42) are significantly increased in CD26 (DPP-4)-/- knockout mice, as illustrated in Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc Natl Acad Sci U S A 2000 97(12):6874-6879.
Accordingly, analysis of GIP in tissues or in the circulation with a C-terminal assay alone may lead to an overestimation of the concentration of full length bioactive GIP (1-42). The majority of circulating GIP immunoreactivity in both the fasting and postprandial states corresponds to the biologically inactive GIP (3-42). GIP Infused into human subjects is rapidly degraded, with a t1/2 of ~7.2 minutes as estimated using N-terminal-specific assays Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab. 2000 Oct;85(10):3575-81.
GIP physiology has also been examined using peptide antagonists, antisera against the GIP receptor, and GIP Receptor-/- mice for "removal" or attenuation of GIP biological activity as outlined in GIP antagonists.
To review the data on GIP and human diabetes, see GIP and human diabetes
GIP and the islet β-cell
GIP stimulates b-cell proliferation and exert anti-apoptotic actions in vitro. Studies using INS-1 b-cells have shown stimulation of b-cell proliferation and enhanced b-cell survival; See Glucose-Dependent Insulinotropic Polypeptide Is a Growth Factor for beta (INS-1) Cells by Pleiotropic Signaling. Mol Endocrinol. 2001 Sep;15(9):1559-70. and Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J Endocrinol. 2002 Aug;174(2):233-46. and Glucose-dependent insulinotropic polypeptide promotes beta-(INS-1) cell survival via cyclic adenosine monophosphate-mediated caspase-3 inhibition and regulation of p38 mitogen-activated protein kinase. Endocrinology. 2003 Oct;144(10):4433-45
Campbell and colleagues demonstrated robust prosurvival actions of GIP on murine and human islet beta cells. Selective conditional elimination of the GIPR in murine beta cells resulted in reduced meal-stimulated insulin secretion under regular chow conditions, decreased accumulation of WAT, yet enhanced sensitivtity to apoptotic injury. High fat feeding reversed the meal-related insulin deficiency in mice with beta cell ablation of the GIPR. RNASeq revealed marked downregulation of Tcf7 expression (a gene related to but distinct from Tcf7L2) in GIPR-deficient islets. The survival of islet beta cells is mediated through novel mechanisms requiring Tcf7, which encodes the Tcf1 protein. Knockdown of Tcf7 in rodent islet cells or TCF7 in human islets increased the susceptbility to apoptosis whereas activation of GIPR signaling increased Tcf7 and decreased apoptosis. Remarkably, Tcf7-/- mice also exhibit marked basal impairment of beta cell function and significantly enhanced sensitivity to islet dysdunction following high fat feeding. These findings reveal a GIPR-Tcf7 axis for control of beta cell function and survival. TCF1 links GIPR signaling to the control of beta cell function and survival Nature Medicine(2015)doi:10.1038/nm.3997
Maida and colleagues compared the actions of a degradation-resistant GIP analogue, with those of exendin-4 or the DPP-4 inhibitor sitagliptin, in murine models of b-cell apoptosis or regeneration in the context of streptozotocin administration. Exendin-4 promoted robust expansion of b-cell mass after STZ, and also reduced the extent of b-cell apoptosis. Sitagliptin had very little effect on b-cell apoptosis but did increase b-cell mass after chronic administration. In contrast, d[Ala2]-GIP had only a modest anti-apoptotic effect and was unable to increase b-cell mass in chronic studies. Furthermore, Gipr-/- mice did not exhibit enhanced susceptibility to STZ-induced b-cell apoptosis in vivo. Hence, the GLP-1 receptor appears to be consistently linked to b-cell proliferation and survival pathways in vivo, whereas the GIP receptor promotes b-cell survival in vitro, but whether GIP exerts significant prosurvival effects in vivo under hyperglycemic conditions remain uncertain. See Differential importance of GIP vs. GLP-1 receptor signaling for beta-cell survival in mice. Gastroenterology. 2009 Dec;137(6):2146-57.
Widenmeir and colleagues demonstrated potent prosurvival actions using a truncated GIP analog, D-Ala(2)-GIP(1-30), D-GIP(1-30), in rat models of T2DM and INS-1 b-cells. Treatment with D-GIP(1-30) produced salutary effects on b-cell mas sin multiple models, with reduced levels of islet pro-apoptotic proteins observed in Vancouver diabetic fatty rats and preserved beta-cell mass noted in streptozotocin-treated rats and Zucker diabetic fatty rats. Studies using cells in vitro demonstrated that D-GIP(1-30) exhibited equivalent insulinotropic and anti-apoptotic potency to GIP(1-42). A GIP receptor agonist exhibits beta-cell anti-apoptotic actions in rat models of diabetes resulting in improved beta-cell function and glycemic control PLoS One. 2010 Mar 9;5(3):e9590
The anti-apoptotic actions of GIP on the islet β-cell in vitro were examined in INS-1(832/13) β cells, subjected to glucoliptocicity. GIP increased Akt, and restored cytoplasmic localization of Foxo1, in association with increased Foxo1 phsophorylation, leading to reduction of Bax expression. Similar findings were observed in primary islet cultures prepared from C57BL/6 mice. Furthermore, a 2 week treatment of Vancouver ZDF rats with intravenous GIP decreased Bax, and upregulated Bcl-2 in β-cells, as described in GIP stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3-K)/ protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1 and downregulation of bax expression. J Biol Chem. 2005 Apr 6; [Epub ahead of print]
Kim and colleagues examined the importance of delayed rectifier channels for the prosurvival effects of GIP in b-cells. Attenuation of channel conductance with TEA reduced INS-1 cell apoptosis following exposure to thapsigargin or staurosporine, whereas cell death was significantly increased following overexpression of Kv2.1 and Kv 1.5 in INS-1 cells. Both GIP and GLP-1 reduced apoptosis induced by thapsigargin. Knockdown of Kv2.1 reduced the extent of thapsigargin-generated apoptosis. Both GLP-1 and GIP also increased phosphorylation and acetylation of Kv2.1 whereas the N-terminally truncated incretins had no effect. Attenuation of cAMP and PKA activity or inhibition of histone acetyltransferase markedly reduced the ability of incretins to phosphorylate and acetylate Kv2.1 and enhance cell survival, whereas the HDAC inhibitor trichostatin potentiated Kv2.1 acetylation but had no effect on Kv2.1 phosphorylation or b-cell survival. Both GIP and GLP-1 promoted cytoplasmic localization of CREB Binding Protein (CBP). RNA interference reduced nuclear CBP expression, and markedly impaired the ability of GIP or GLP-1 to lysine acetylate Kv2.1. GIP and GLP-1 also promoted internalization of Kv2.1 in human islets. See Pancreatic β-cell prosurvival effects of the incretin hormones involve post-translational modification of Kv2.1 delayed rectifier channels Cell Death Differ. 2011 Aug 5. doi: 10.1038/cdd.2011.102
The actions of GIP to activate Akt appear to be mediated through unique pathways independent of PI3K. Both GIP and GLP-1 stimulated Akt activation in kinase assays and promoted b-cell survival (INS-1 cells and mouse islets) through pathways that likely involved cAMP but not phosphorylation of T308 or S473. See Non-canonical activation of Akt/PKB in beta -cells by the incretin hormone glucose-dependent insulinotropic polypeptide (GIP). J Biol Chem. 2009 Feb 20.
Consistent with the differences seen comparing GIP vs. GLP-1 on the β-cell secretion response in experimental and clinical diabetes, pre-clinical studies have illustrated differences in the b-cell signaling molecules triggered by these two related peptides. Intriguingly, although SUR-/- mice exhibit a defect in the response to GLP-1 despite an increase in the levels of cyclic AMP in SUR-/- islets Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002 Oct 4;277(40):37176-83, In contrast, mice with a targeted inactivation of the Kir6.2 gene retain an insulinotropic response to GLP-1, but not GIP, as illustrated in Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like Peptide-1 on insulin secretion and gut motility. Diabetes. 2005 Apr;54(4):1056-63
GIP and blood vessels
GIPR expression has been localized to blood vessels, predominantly endothelial cells, where it activates signal transduction pathways, increases endothelin-1 (ET-1) secretion and stimulates cell proliferation in cell culture Glucose-dependent insulinotropic peptide stimulates thymidine incorporation in endothelial cells: role of endothelin-1 Am J Physiol Endocrinol Metab. 2003 Aug;285(2):E390-6, however an important physiological role for GIP in ECs has not yet been demonstrated. Berglund and colleagues localized GIPR expression predominantly to mouse aortic endothelial cells, and to a lesser extent, in smooth muscle cells by immunocytochemistry. Culture of mouse aorta ex vivo for 3 days increased osteopontin synthesis and secretion and enhanced ET-1 release; ET-1 secreted from EC cells in turn increased osteopontin secretion from VSMCs. ET-1 antagonists blocked the GIP-dependent induction of osteopontin secretion from vascular smooth muscle cells. The GIPR-dependent increase in ET-1 and ET-1-dependent induction of OPN were blocked by a small molecule CREB antagonist, but GIP did not increse cAMP in the same experiments. Expression of the human GIPR and OPN mRNAs were positively correlated in atherosclerotic plaques. Patients with type 2 diabetes carrying the SNP rs10423928 in the GIPR had an increased risk of stroke. GIP infusion in non-diabetic subjects increased plasma OPN levels in a genotype ( (TA/AA)-dependent manner Glucose-Dependent Insulinotropic Polypeptide (GIP) Stimulates Osteopontin Expression in the Vasculature via Endothelin-1 and CREB Diabetes doi:10.2337/db15-0122
For an overview of GIP, GLP-1 and GLP-2 actions on blood vessels, see Vascular Biology of Glucagon Receptor Superfamily Peptides: Mechanistic and Clinical Relevance Endocr Rev. 2016 Dec;37(6):554-583
GIP, adipocytes and obesity
GIP receptors are expressed on adipocytes and may transduce a large number of actions on adipocyte biology, as reviewed in GIP biology and fat metabolism. Life Sci. 2000;66(2):91-103. Experimental data derived from studies of the GIP receptor knockout mouse strongly implicates a role for the GIP receptor in the regulation of body weight. The Seino lab has shown that GIPR-/- mice are resistant to the development of diet-induced obesity. Furthermore, GIPR-deficient ob/ob double mutant mice exhibited a 41% reduction in body weight compared to normal ob/ob mice and plasma lipids such as triglycerides, free fatty acids, and cholesterols were lower in GIPR-deficient ob/ob mice than in normal ob/ob mice. Moreover, energy expenditure was increased following high fat feeding in mice with absent GIPR signaling, indicating that inhibition of the GIP signal ameliorates obesity and obesity-related hyperglycemia, and dyslipidemia. These findings are described in Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002 Jul;8(7):738-42.
GIP may also be important for regulation of adipokine secretion. Gipr-/- mice exhibit reduced adipocyte mass and defective upregulation of PAI-1 and resistin following high fat feeding. Furthermore, GIP, but not exendin-4, upregulates levels of plasma resistin in acute or chronic experiments, as outlined in Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest. 2007 Jan 2;117(1):143-152. The stimulation of resistin secretion appears to be direct, as GIP, in the presence of insulin, increased resistin secretion through a pathway involving p38 mitogen activated protein kinase (p38 MAPK) and the stress-activated protein kinase/Jun-amino-terminal kinase (SAPK/JNK) in differentiated 3T3L1 adipocytes. Intriguingly, resistin mimics many of the molecular aspects of GIP action on adipocytes in vitro, as shown in Resistin is a key mediator of glucose-dependent insulinotropic polypeptide (GIP) stimulation of lipoprotein lipase (LPL) activity in adipocytes. J Biol Chem. 2007 Sep 20; [Epub ahead of print].
Remarkably, many of the actions of GIP on murine adipocytes were completely lost in resistin-/- and even in resistin+/- heterozygote mice. GIP plus insulin had no effect on LPL activity, pAkt, pAMPK, or fatty acid uptake in resistin-/_ or +/- adipocytes. Furthermore, GIPR protein expression was markedly downregulated in WAT, BAT, and the SVF fraction, even in Retn+/- mice whereas 100 nM resistin increased GIPR expression after 72 hrs in Retn+/+ adipocytes. The induction of GIPR expression by resistin was demonstrated to be via transcriptional induction via c-Jun and the SAPK/JNK pathway Resistin Knockout Mice Exhibit Impaired Adipocyte Glucose-Dependent Insulinotropic Polypeptide Receptor (GIPR) Expression Diabetes. 2012 Sep 21
GIP promotes induction of LPL activity and triglyceride (TG) accumulation in differentiated insulin-treated 3T3-L1 adipocytes, in association with phosphorylation of protein kinase B (PKB) and reductions in phosphorylated LKB1 and AMP-activated protein kinase (AMPK). Similar findings were observed in experiments with human adipocytes. Moreover, treatment of Vancouver Diabetic Fatty (VDF) Zucker rats with GIP led to increased LPL activity and triglyceride accumulation in adipose tissue-See Activation of lipoprotein lipase (LPL) by glucose-dependent insulinotropic polypeptide (GIP) in adipocytes: A role for a protein kinase B (PKB), LKB1 and AMP-activated protein kinase (AMPK) cascade. J Biol Chem. 2007 Mar 23;282(12):8557-67.
The extent to which GIP is important for lipid metabolism in humans is less clear. GIP consistently exerts potent insulinotropic actions when administered acutely to non-diabetic subjects however acute GIP administration has little effect on meal-related gastric emptying or plasma lipid levels in the presence or absence of intravenous intralipid administration in non-diabetic subjects On the role of glucose-dependent insulintropic polypeptide, GIP, in postprandial metabolism in humans. Am J Physiol Endocrinol Metab. 2010 Mar;298(3):E614-21
GIP also promotes calcitonin (CT) and CGRP gene expression in human adipocytes in vitro. Procalcitonin levels are increased in many subjects with obesity and insulin resistance, and some of these subjects alsio exhibit increases in circulating GIP. Adipocyte differentiation was associated with marked upregulation of Gipr expression, and exogenous GIP stimulated cAMP formation in human adicpoytes in vitro. GIP rapidly increased CT and CGRP gene expression by 1 hr (through mechanisms sensitive to the PKA inhibitor H89), and increased CGRP-1 protein (but not procalcitonin) was detected in the cell culture medium following GIP treatment. See Glucose-Dependent Insulinotropic Polypeptide (GIP) Induces Calcitonin Gene-Related Peptide (CGRP)-I and Procalcitonin (Pro-CT) Production in Human Adipocytes J Clin Endocrinol Metab. 2010 Nov 24. [Epub ahead of print]
The effects of GIPR activation on cytokine expression and lipolysis in human adipocytes have also been examined in vitro. Adipocytes were prepared from subcutaneous abdominal fat depots from 9 subjects (4 non-obese). Stromal vascular cells were expanded in vitro, then differentiated with a cocktail that included T3, dexamethasone, insulin, rosiglitazone, vitamins, and transferrin. Treatment of cells for 1h with 1 nM GIP rapidly induced cytokine (IL-6, IL-1β and IL-1Ra) mRNA expression in the presence or absence of serum, actions sensitive to PKA, orlistat (lipase inhibitor) or IKK-β inhibition . These actions were blocked by treatment of cells with GIP(6-30) and Gipr mRNA transcripts were not detected in undifferenitaed pre-adipocytes. In contrast, GIP treatment had little effect on cytokine secretion, but potentiated the induction of cytokine secretion in the presence of other inflammatory mediators (LPS, IL-1b, TNF-a). Consistent with the inhibitory actions of orlistat on cytokine gene expression, GIP induced lipolysis as measured by glycerol release, in a PKA-dependent manner. Inhibitors of IKK-β/NF-κB, IL-1 receptor activation, Jun N-terminal kinase (JNK), and the Mitogen-Activated Protein Kinase (MAPK) blocked GIP-induced HSL phosphorylation and lipolysis. Acute GIP treatment alone, or with insulin, did not affect glucose uptake, whereas prolonged pretreatment with GIP for 6 h dose-dependently reduced insulin stimulated glucose uptake, in association with impaired insulin-stimulated GLUT4 translocation Glucose-dependent insulinotropic polypeptide induces cytokine expression, lipolysis and insulin resistance in human adipocytes Am J Physiol Endocrinol Metab. 2012 Oct 23.
The effects of a 4 hr GIP infusion in human subjects, with or without accompanying insulin clamps, as well as the effects of GIP administration on macrophage and adipocyte cultures ex vivo, revealed that GIP infusion increased plasma levels of MCP-1, independent of levels of glucose or insulin, in obese (BMI >28) healthy male volunteers. Transcriptomic analysis of gene expression in RNA from human subcutaneous adipose tissue biopsies revealed increased mRNA levels for MCP-1 and MCP-2, IL-6, and CD-68. GIP plus diprotin A stimulated cyclic AMP accumulation in monocyte/macrophage cell lines and primary human peripheral blood monocytes but GIP alone did not alter p42/p44 ERK phosphorylation whereas stimulation was observed with addition of diprotin A. GIP alone had no effect on stimulation of cytokine expression in multiple macrophage cell lines or human adipocytes- Simpson–Golabi–Behmel syndrome (SGBS preadipocytes) or differentiated NIH-3T3-L1 cells, however co-culture of 3T3-L1 cells and RAW264 macrophages enabled GIP-dependent induction of MCP-1 mRNA transcripts. Using a cytokine array to assess secreted proteins, Mip-1b levels were increased by GIP from RAW cells but not from NIH 3T3-L1 cells. GIP increases adipose tissue expression and blood levels of MCP-1 in humans and links high energy diets to inflammation: a randomised trial Diabetologia. 2015 May 21
Varol and colleagues examined the consequences of GIPR activation on adipose tissue and systemic inflammation in HF fed mice treated with d[Ala2]-GIP, 0.12 mg/g, once daily for 10-14 weeks. Although adipocyte size (more than 2-fold increase in total adipocyte area) and WAT mass was increased, proinflammatory macrophage infiltration (CD11c+ ATMs, together with reduced infiltration of IFNg+ CD8+ and CD4+ T cells.) and levels of inflammatory cytokines were reduced in epididymal adipose tissue of high fat fed mice, along with increased insulin sensitivity. Upregulated expression of mRNA transcripts encoding for lipid droplet proteins PLIN1, CIDEA, and CIDEC, as well as their transcription factor regulator, PPARg, was also detected in WAT. A significant reduction in the number of both CD8+ and CD4+ T cell populations was detected in WAT, and reduced levels of circulating monocytes and neutrophils was observed in mice that received [d-Ala2]GIP. [d-Ala2]GIP of human WAT explants ex vivo for 24 h also reduced the expression of inflammatory cytokines and chemokines. Long-Acting Glucose-Dependent Insulinotropic Polypeptide Ameliorates Obesity-Induced Adipose Tissue Inflammation J Immunol. 2014 Sep 12. pii: 1401149
The insulin-sensitizing actions of GIP were examined in differentiated 3T3-L1 cells. Although GIP alone had little effect on glucose uptake or insulin signaling pathways, GIP potentiated the effect of insulin to enhance GLUT4 translocation, glucose uptake, nuclear Fox01 exclusion. The effect of GIP was somewhat delayed, and required at least 30 minutes of pre-exposure to GIP, prior to insulin administration. Pharmacological inhibition of Akt or Akt2 or rab10 knockdown impaired the potentiating actions of GIP, however GIP alone did not activate Akt phosphorylation, nor effect insulin receptor number, binding or phosphorylation. Increased levels of cAMP did not mimic the insulin sensitizing effects of GIP, however inhibition of cAMP or PKA action reduced some but not all of the effects of GIP on insulin action in 3T3-L1 cells. Exposure of cells to TGX-221, an inhibitor ofp110gamma abolished the ability of GIP to potentiate insulin-stimulated GLUT4 translocation Gastric inhibitory peptide controls adipose insulin sensitivity via activation of CREB and p110β isoform of PI3 kinase J Biol Chem. 2011 Oct 25.
A link between GIP action and induction of osteopontin expression has also been proposed. Ahlqvist detected expression of an immunoreactive GIPR protein in adipose tissue and muscle using an antibody (Kieffer lab). Gipr mRNA transcripts were also detected in visceral and subcutaneous adipose tisue from human subjects undergoing gastric bypass. Although plasma GIP levels were increased in these obese subjects, adipose tissue Gipr mRNA transcripts tended to be inversely correlated with BMI, waist circumference and visceral fat mass. Increased adipose Gipr expression correlated with increased insulin sensitivity. Different Gipr variant alleles exhibit different correlations with BMI, fat mass, and insulin sensitivity in human subjects. Over 60 Gipr splice variants were detected by RT-PCR in human adipose tissue, of which only 2 corresponded to full length Gipr mRNAs. GIP stimulated osteopontin gene expression in isolated rat adipocytes in the presence of insulin(1 nM) and adipocyte osteopontin gene epression correlated with insulin resistance. A link between GIP and osteopontin in adipose tissue and insulin resistance Diabetes. 2013 Jan 24
GIP and the central nervous system
Although very little is known about the role of GIP in the brain, GIP was found to be expressed in the rodent hippocampus and the extent of GIP expression correlated with the rate of cell proliferation in the experimental model system. Furthermore, the GIP receptor was also expressed in the dentate gyrus, and activation of GIP receptor signaling stimulated cell proliferation, whereas adult GIP receptor knock-out mice exhibit a significantly lower number of newborn cells in the hippocampal DG compared with wild-type mice Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J Neurosci. 2005 Feb 16;25(7):1816-25
GIP may also exert actions in the peripheral nervous system. Both GIP and GIPR RNA were detectable in peripheral nerves and sciatic root ganglia and in sections from spinal cord, and GIP and GIPR RNA transcripts were transiently upregulated in the spinal cord following peripheral nerve crush injury. Immunocytochemical studies demonstrated localization of GIP and GIPR immunopositivity to distinct populations of cells within the rat spinal cord, including neurons, Schwann cells and oligodendrocytes but not astrocytes. Functionally, spontaneous nerve generation was impaired in Gipr-/- mice following crush injury as outlined in Glucose-dependent insulinotropic polypeptide (GIP) and its receptor (GIPR): Cellular localization, lesion-affected expression, and impaired regenerative axonal growth J Neurosci Res. 2009 Jan 23. [Epub ahead of print]
Killion and colleagues examined the consequences of GIPR blockade alone, or in combination with GLP-1R agonism in mice and non-human primates. Peripheral GIPR blockade protected agaonst DIO in mice but did not itself result in weight loss, whereas monkeys treated with the antibody did lose weight. In these studies, the GIPR blocking antibidy sensitized animals to co-administration of GLP-1R agonists. Hence these studies argue for combined GIPR blockade/GLP-1R agonism to treat obesity. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models Sci Transl Med. 2018 Dec 19;10(472)
Kaneko and colleagues identified a role for central GIPR signaling in the control of food intake and leptin sensitivioty. GLP1R Administration of GIP into the CNS impaired leptin action, whereas reduction of the CNS GIPR activity using a blocking antibody enhanced leptin sensitivity and reduced food intake in obese but not in lean mice. Peripheral administration of a GIPR agonist impaired acute leptin sensitivity. Peripheral administration of the Gipg013 antibody did not reduce body weight but prevented further weight gain in HFD-fed mice. Moreover, the central blockade of GIPR with the Gipg013 antibody did not sensitize mice to the weight loss effects of the GLP-1R agonist liraglutide. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition J Clin Invest. 2019 Aug 12;130:3786-3791.
Using a reporter mouse generated by crossing Gipr-Cre mice with a ROSA26-EYFP line, putative GIPR+ cells were detected within mouse olfactory bulb, cortical layers, hippocampus, the medial preoptic area, subfornical organ, anterodorsal thalamic nucleus, magnocellular preoptic nucleus, paraventricular thalamic nucleus, suprachiasmatic nucleus, as well as the interfascicular nucleus. Most GIPR+ cells also expressed SST as determined by signle cell RNASeq. Of possible relevance to control of feeding GIPR+ cells wer also localized to the paraventricular, dorsomedial, and arcuate nuclei of the hypothalamus and the area postrema. A subset of mouse and human hypothalamic neurons was reported to express both the GIPR and GLP1R. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake Cell Metab. 2019 Nov 5;30(5):987-996.e6. It remains unclear whether the GIP gene is meaningfully expressed within the CNS, or if GIP simply enters the brain via passive or active transport. Functionally, chemogenetic activation of murine hypothalamic Gipr cells resulted in acute reduction in food intake. Nevertheless, use of chemogenetics alone to simultaneously activate GIPR and GLP1R, or administration of submaximal doses of peripheral exendin-4 together with GIPR chemogenetic activation, did not reveal evidence for additive or synergistic reduction of food intake Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake Cell Metab. 2019 Nov 5;30(5):987-996.e6.
GIP and the HPA axis
The GIP receptor is expressed in cells within the hypothalamus, pituitary, and adrenal cortex, in rodents. In humans, aberrant expression of the GIP receptor in adrenal adenomas produces increased cortisol levels pursuant to meal ingestion, leading to meal-induced Cushings syndrome Gastric inhibitory polypeptide-dependent cortisol hypersecretion--a new cause of Cushing's syndrome N Engl J Med. 1992 Oct 1;327(14):974-80 and Ectopic and abnormal hormone receptors in adrenal Cushing's syndrome Endocr Rev. 2001 Feb;22(1):75-110. As Gipr-/- mice exhibit a lean phenotype with resistance to diet-induced obesity, Bates and colleagues assessed the role of the adrenal GIPR and corticosterone in the Gipr-/- phenotype. GIPR activation increased corticosterone secretion from WT mice in a rapid dose-dependent manner, in both normoglycemic and diabetic mice, whereas Gipr-/- mice exhibited significant reductions in ambient levels of plasma corticosterone. GIP directly stimulated corticosterone secretion from adrenal cells, and activated an adrenal gene expression program promoting corticosterone biosynthesis. Gipr-/- mice exhibited enhanced sensitivity to ACTH, manifested by upregulation of adrenal ACTH receptor expression, and greater corticosterone secretion in response to ACTH ex vivo. Nevertheless, corticosterone supplementation did not modify the principal metabolic phenotypes of high fat fed Gipr-/- mice. Gipr Is Essential for Adrenocortical Steroidogenesis; However, Corticosterone Deficiency Does Not Mediate the Favorable Metabolic Phenotype of Gipr−/− Mice Diabetes published ahead of print October 31, 2011, doi:10.2337/db11-1060
GIP and bone
GIP is anabolic for bone and appears to exert direct actions on osteoblasts as GIPR mRNA and protein are expressed in normal bone and osteoblast-like cell lines. GIP stimulates increases in cAMP and intracellular Ca2+ levels in cultured osteoblasts and these effects are coupled to markers of new bone formation, including elevations in alkaline phosphatase activity and collagen type 1 mRNA as outlined in Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology 2000;141:1228-35. GIP also increases bone mineral density in ovariectomized rats, a rodent model of postmenopausal osteoporosis Glucose-dependent insulinotropic peptide is an integrative hormone with osteotropic effects. Mol Cell Endocrinol 2001;177:35-41. Relative to age-matched wild type mice, younger Gipr-/- mice exhibit reduced bone size and mass, abnormal bone microarchitecture, impaired biomechanical properties and altered bone turnover Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone 2005;37:759-69 . However, as the mice age, these differences become less apparent. The presence of GIPR mRNA and protein has also recently been detected in rodent osteoclast cells and GIP administration was found to have inhibitory effects on bone resorption Effects of glucose-dependent insulinotropic peptide on osteoclast function. Am J Physiol Endocrinol Metab. 2007 Feb;292(2):E543-. Moreover, adult Gipr-/- mice exhibit reductions in parameters of bone formation, including altered bone size and bone mass as well as increases in plasma Ca2+ levels following meal ingestion, and increased numbers of osteoclasts associated with enhanced bone turnover, suggesting that GIP may provide a direct link between Ca2+ from a meal and calcium deposition in bone Glucose-dependent insulinotropic polypeptide receptor knockout mice have altered bone turnover. Bone. 2005 Dec;37(6):759-69 and Gastric inhibitory polypeptide as an endogenous factor promoting new bone formation after food ingestion. Mol Endocrinol. 2006 Jul;20(7):1644-51. Transgenic expression of GIP also increased osteoblastic activity in older mice and prevents the age-associated decline in bone mass and bone strength in mice. GIP recepotor expression that declined with age was observed in bone marrow stromal cells (BMSC), a potential osteoblastic precursor population. Impact of glucose-dependent insulinotropic peptide on age-induced bone loss. J Bone Miner Res. 2008 Apr;23(4):536-43.
These studies implicate an important and novel role for GIP in the regulation of bone remodeling. However, acute administration of GIP does not alter markers of bone turnover in human studies Role of gastrointestinal hormones in postprandial reduction of bone resorption. J Bone Miner Res. 2003 Dec;18(12):2180-9, and whether more long-term application of GIP will modulate bone turnover in humans is not known.
Acute infision of native GIP for 12 minutes also suppressed markers of bone resorption under conditions of normal of elevated (12 mM) glucise in human subjecs with type 1 diabetes Glucose-Dependent Insulinotropic Polypeptide (GIP) Inhibits Bone Resorption Independently of Insulin and Glycemia J Clin Endocrinol Metab. 2018 Jan 1;103(1):288-294
GIP and the Immune System
TheGIPreceptor is expressed at low levels in bon marrow-derived cells, principally in the myeloid lineage. Loss of the Gipr in Gipr-/- mice leads to impaired hematopoeisis, including a deficiency of circulating neutrophils and reduced levels of Notch ligand expression in osteoblast-enriched bone marrow extracts Glucose-Dependent Insulinotropic Polypeptide Receptor Deficiency Leads to Impaired Bone Marrow Hematopoiesis J Immunol 2017 Apr 15;198(8):3089-3098.
The importance of basal GIPR signaling for suppression of inflammation was further revealed in studies of HFD-fed mice with complete whole body or myeloid GIPR deficiency. GIP was shown to suppress macrophage inflammation in adipose tissue, whereas selective depletion of the bone marrow GIPR or myeloid GIPR augmented expression of the alarmin S100!8/9, associated with increased adipose tissue inflammation and increased adipose tissue browning. GIP regulates inflammation and body weight by restraining myeloid-cell-derived S100A8/A9 Nat Metab. 2019 Jan;1(1):58-69
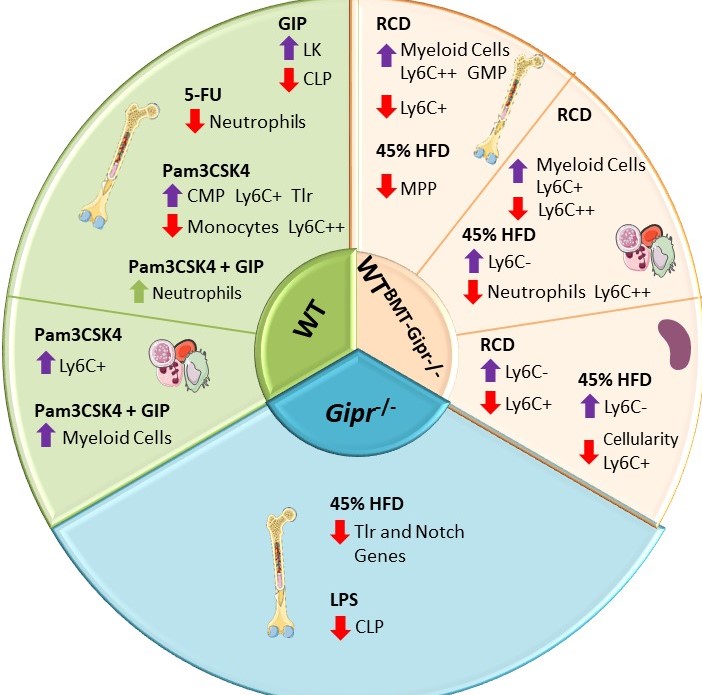
Gemma Pujadas examined the importance of the bone marrow GIPR for the response to proinflammatory stress and stress-related hematopoiesis. Gipr was identified in T cells, myeloid cells, and myeloid precursors, however proportions of these cell populations were not different in peripheral blood, spleen, or BM of Gipr-/- and GiprTie2-/- mice. GIPR signaling controls the expression of BM Toll-like receptor (TLR) and Notch-related genes regulating hematopoiesis. Consistent with the data from Mantelmacher et al, loss of the BM GIPR attenuated the extent of adipose tissue inflammation and dysregulated the hematopoietic response to BMT. GIPR agonism modified BM gene expression profiles following 5-FU and Pam3CSK4 whereas loss of the Gipr altered the hematopoietic responses to energy excess, and two TLR ligands, and 5-FU. Nevertheless, the magnitude of these GIPR-associated changes was comparatively modest. The gut hormone receptor GIPR links energy availability to the control of hematopoiesis Mol Metab. 2020 Sep;39:101008